Allosterische Modulation nikotinischer Acetylcholinrezeptoren – eine neue Therapieoption für die Behandlung von -Nervenkampfstoffvergiftungen?
Aus dem Institut für Pharmakologie und Toxikologie der Bundeswehr, München 1 (Leiter: Oberstarzt Prof. Dr. H. Thiermann) und dem Department Pharmazie – Zentrum für Pharmaforschung der Ludwig-Maximilians-Universität, Lehrstuhl für Pharmazeutische / Medizinische Chemie, München2 (Prof. Dr. K.T. Wanner)
Zusammenfassung
Hochtoxische phosphororganische Verbindungen, seien es Nervenkampfstoffe oder bestimmte Pestizide, begründen ihre Toxizität durch die irreversible Hemmung der Acetylcholinesterase (AChE), dem Schlüsselenzym im Prozess der Neurotransmission. Da die katalytische Hydrolyse des Acetylcholins ausbleibt, und somit der intrinsische Effekt von Acetylcholin nicht mehr terminiert wird, akkumuliert dieser Neurotransmitter unkontrolliert im synaptischen Spalt. Infolgedessen ist die Funktion der nikotinischen (nAChR) und muskarinischen Acetylcholinrezeptoren (mAChR) wegen der Überstimulation und insbesondere durch die Desensitisierung der nAChR beeinträchtigt.
Das resultierende cholinerge Syndrom wird lebensbedrohlich, wenn keine adäquate Therapie erfolgt. Die derzeitige Standardtherapie, bestehend aus einem kompetitiven mAChR-Antagonisten (z. B. Atropin) und einem Oxim zur Reaktivierung der AChE (z. B. Obidoxim, Pra-lidoxim, HI-6), ist nicht ausreichend bei Vergiftungen mit Soman oder Tabun. Daher sind alternative therapeutische Optionen dringend notwendig. Ein innovativer Ansatz beinhaltet den Einsatz von Wirkstoffen, die selektiv mit den nAChR interagieren. Genauer gesagt, könnte der Einsatz von positiv allosterischen Modulatoren (PAM), welche die Population des offenen und im Prinzip metastabilen Zustands der nAChR erhöhen, vielversprechend sein. MB327 (1,1’-(Propan-1,3-diyl)bis(4-tert-butylpyridinium)diiodid) ist in der Lage, die Muskelkraft in Gewebepräparationen verschiedener Spezies (inklusiv humaner Atemmuskelpräparate) nach Somanvergiftung wieder herzustellen. Kürzlich konnte aufgeklärt werden, dass die Wirkweise von MB327 rezeptorvermittelt erfolgt: Die Aktivität vormals desensitisierter nAChR konnte wieder hergestellt werden, eine typische Eigenschaft von sogenannten Typ II PAM. Allerdings zeigt MB327 keine hohe Effektivität und Selektivität. Folglich sind effizientere und innovative Wirkstoffe, die auf dem optimalen Pharmakophor basieren, zu entwickeln.
Für die Entwicklung neuer Antidote, die insbesondere bei der Therapie von Soman- oder Tabun-Vergiftungen eingesetzt werden können, ist die Aufklärung von Struktur-Wirkungsbeziehungen erforderlich. Da erste Ergebnisse vielversprechend sind, sollte diese Herausforderung mit Nachdruck angenommen werden.
Stichworte:
Nervenkampfstoffe, Vergiftung, therapeutischer Ansatz, ni-ko-tinische Acetylcholinrezeptoren, Desensitisierung, positiv allosterische Modulatoren, pharmakologische Untersuchungen, Wirkstoffe, Drug Design
Keywords:
Nerve agents, poisoning, therapeutic approach, nicotinic acetylcholine receptors, desensitisation, positive allosteric modulators, pharmacological investigations, drugs, drug design
Einführung
Nicht zuletzt der Einsatz von Nervenkampfstoffen gegen die Zivilbevölkerung Anfang April 2017 in Syrien verdeutlicht, dass chemische Kampfstoffe immer noch eine ernsthafte Bedrohung sowohl für militärisches Personal als auch die Zivilbevölkerung sind. Insbesondere die relativ leichte Herstellung hochtoxischer phosphororganischer Verbindungen, seien es Nervenkampfstoffe oder bestimmte Pestizide, unterstreicht die Notwendigkeit einer effektiven medizinischen Behandlung.
Der entscheidende toxische Mechanismus phosphororganischer Verbindungen beruht auf der Inaktivierung der Acetylcholinesterase (AChE, EC 3.1.1.7), einer Serinhydrolase, aufgrund von Phosphorylierung bzw. Phosphonylierung der Hydroxylgruppe des Serins, das im aktiven Zentrum des Enzyms lokalisiert ist [1, 2]. Die AChE terminiert die intrinsische Aktivität des Neurotransmitters an der postsynaptischen Membran im Neuron bzw. der neuromuskulären Endplatte. Ist die AChE inhibiert, wird der hydrolytische Abbau des Acetylcholins nicht mehr katalysiert. Da Acetylcholin aus der Präsynapse weiterhin freigesetzt wird, kommt es zur Anreicherung des Neurotransmitters im synaptischen Spalt [3] und zur eingeschränkten Funktion cholinerger Rezeptoren, einerseits muskarinischer (mAChR), andererseits nikotinischer Acetylcholinrezeptoren (nAChR).
Das resultierende cholinerge Syndrom äußert sich mit periphernervösen (z. B. Bronchorrhoe, Bronchospasmen, veränderten Herzfrequenzen, muskulären Paralysen, unkontrolliertem Urinieren und Defäkation) und zentralnervösen Effekten (z. B. Zittern, Krämpfe, Atemstillstand) [4,5]. Der Tod tritt letztendlich durch peripheren und zentralen Atemstillstand ein [6, 7].
Derzeitige Therapie von Nervenkampfstoffvergiftungen
Da das Syndrom rasch fortschreitet und die Lähmung der Atemmuskulatur droht, muss die Behandlung sofort eingeleitet werden, in der Regel durch Ersthelfer oder im militärischen Bereich im Rahmen der Selbst- und Kameradenhilfe [8]. Die Standardbehandlung umfasst die Gabe von Atropin, das ein kom-petitiver Antagonist für mAChR ist, und von Oximen (z. B. Obidoxim, Pralidoxim, HI-6), welche die Acetylcholinesterase reaktivieren [9, 10]. Zusätzlich sind noch Antikonvulsiva angebracht, um die cholinerg vermittelten Effekte im zentralen Nervensystem zu mildern [11].
Atropin antagonisiert die durch das akkumulierte Acetylcholin hervorgerufenen Effekte an den mAChR, nicht aber an den nAChR, die insbesondere für die Muskelfunktion entscheidend sind. Die nikotinergen Effekte werden derzeit indirekt behandelt: Oxime dephosphylieren die inhibierte AChE und stellen dadurch deren katalytische Aktivität wieder her. Der resultierende Abbau des überschüssigen Acetylcholins und die Rückkehr in den Normbereich führen dazu, dass sowohl nAChR als auch mAChR wieder „normal“ funktionieren [12]. Bis heute ist jedoch noch kein Breitspektrum-Oxim verfügbar, das universal als einziges Antidot für die Behandlung aller Art von Nervenkampfstoffvergiftungen eingesetzt werden kann [13]. Daher scheint die Kombination mehrerer Oxime mit komplementärer und systematischer Wirkung ein brauchbares Konzept zu sein [14].
Der Einsatz von Oximen ist jedoch limitiert, wenn die AChE nicht mehr oder nur unzureichend reaktivierbar ist. Dies kann daran liegen, dass im menschlichen Körper der Enzym-Nervenkampfstoff-Komplex entweder generell für eine Reaktivierung nicht zugänglich ist, wie im Fall von Tabun [15], oder die Reaktivierung durch nachfolgende chemische Prozesse, wie beispielsweise einer Dealkylierung, verhindert wird. Diese sogenannte „Alterung“ des Enzym-Organophos(phon)at-Komplexes kann bei Soman innerhalb weniger Minuten stattfinden [16 - 18]. In diesen Fällen kann die – durch die nAChR-Dysfunk-tion verursachte – zum Erliegen gekommene neuromuskuläre Transmission nicht medikamentös behoben werden. Der dadurch hervorgerufene periphere Atemstillstand ist ein lebensbedrohliches Problem, das zukünftig mit einem neuen therapeutischen Ansatz behoben werden soll. Konkret ist es das Ziel, die neuromuskuläre Transmission speziell in der Atemmuskulatur wieder herzustellen [19].
Folglich sind Wirkstoffe, die selektiv mit den nAChR interagieren, von therapeutischem Interesse. Insbesondere dann, wenn der endogene cholinerge Tonus trotz des Acetylcholinüberschusses durch diese Wirkstoffe kontrolliert, d. h. per Dosis und Anwendungszeit wieder hergestellt werden kann [20].
Nikotinische Acetylcholinrezeptoren
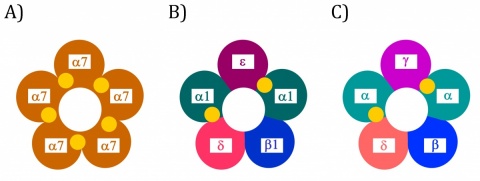
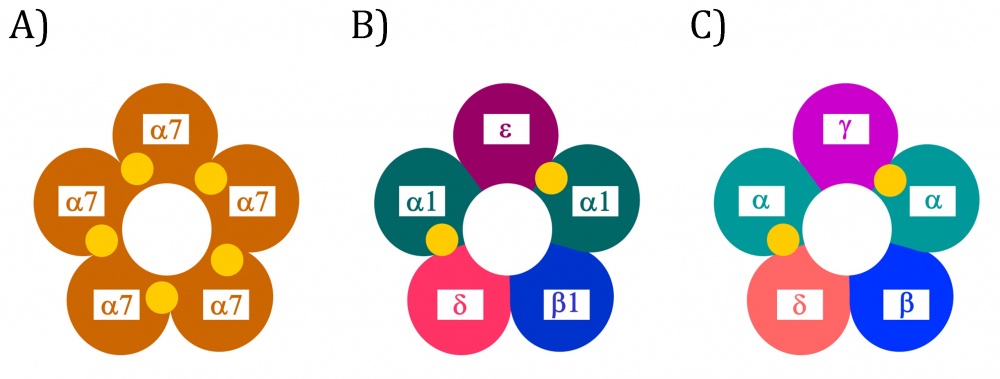
Der nAChR besteht aus fünf Untereinheiten (mit den griechischen Buchstaben α, β, γ, δ und ε bezeichnet), die jeweils aus einer extrazellulären Domäne, einer Transmembranregion und einer intrazellulären Domäne bestehen. Die fünf Untereinheiten sind miteinander nicht kovalent verbunden und können entweder identisch (homomer, z. B. 5α7) oder verschieden (heteromer, z. B. 2α1β1δε) sein [26]. Sie sind so miteinander arrangiert, dass sich in der Mitte eine Pore bildet.
Der sogenannte Muskeltyp-nAChR ist ein heteromerer Subtyp und setzt sich aus den Untereinheiten 2α1β1δε zusammen [27]. Dieser Subtyp ist ausschließlich an der neuromuskulären Synapse lokalisiert und wandelt ein chemisches in ein elektrisches Signal um. In pathophysiologischen Situationen (z. B. Atrophie aufgrund Immobilisation) werden offensichtlich auch homomere 5α7-nAChR im Muskel hochexprimiert, die dann in der Neurotransmission involviert sind; vermutlich handelt es sich um ein Backup-System [28, 29].
Der endogene Agonist Acetylcholin bindet in Bindungstaschen, die sich in der extrazellulären Domäne befinden und jeweils von zwei Untereinheiten gebildet werden. Diese sogenannte orthosterische Bindungsstelle befindet sich bei dem heteromeren Muskeltyp 2α1β1δε-nAChR in dem α/δ- und dem α/ε-Interface. Beim homomeren 5α7-nAChR sind theoretisch 5 -orthosterische Bindungsstellen möglich (jeweils eine pro α/α-Interface), für die Aktivierung reicht aber die Besetzung zweier Bindungsstellen aus [30]. Der nAChR ist relativ hoch konserviert. Beispielsweise weist der Muskeltyp-nAChR des Zitterrochens (2αβγδ) ein hohe Homologie zum humanen Muskeltyp-nAChR (2α1β1δε) auf [31] (Abbildung 1).
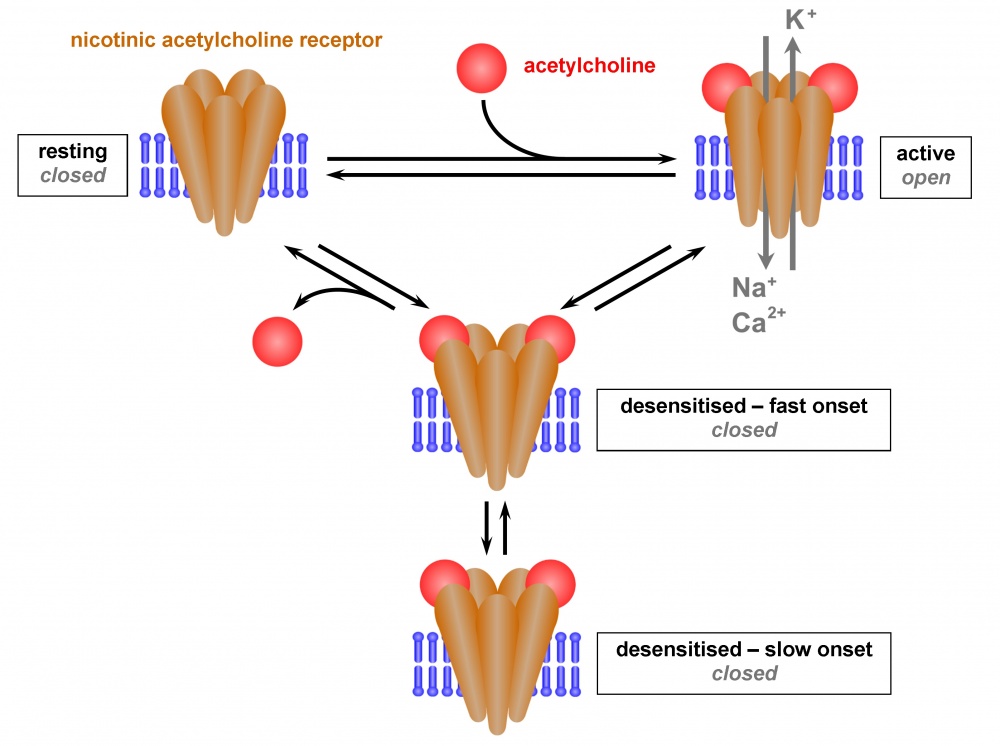
Im geschlossenen Zustand sind die 5 Untereinheiten miteinander verdrillt, so dass sich in der Transmembranregion die engste Stelle befindet. Bindet Acetylcholin an die beiden Bindungsstellen, „entdrillt“ sich der Ionenkanal und erweitert seine Pore [32], wodurch Kationen hindurch geleitet werden: Natrium- und Calciumionen nach innen und Kaliumionen nach außen [33]. Der Natriumeinstrom führt zu einer Depolarisation der postsynaptischen Membran, die ihrerseits weitere Kaskaden in Gang setzt, beim neuromuskulären System letztendlich die Muskelkontraktion [34]. Der nAChR kann auch durch andere Agonisten, wie Carbamoylcholin, Nikotin oder Epibatidin, aktiviert werden, die ebenfalls an die orthosterischen Bindungsstellen binden. Die orthosterischen Bindungsstellen können aber auch durch kompetitive Antagonisten, wie beispielsweise α-Bungarotoxin oder Conotoxine, adressiert werden, so dass der intrinsische Effekt unterbunden wird [35].
Neben den orthosterischen Bindungsstellen sind am nAChR zahlreiche sogenannte allosterische Bindungsstellen vorhanden. Binden dort Liganden, wird die Aktivierung des nAChR entweder in positiver oder negativer Weise beeinflusst. Folglich werden diese Substanzen als positive allosterische Modulatoren (PAM) oder als negative allosterische Modulatoren (NAM) bezeichnet [36].
Aktivierungszustände
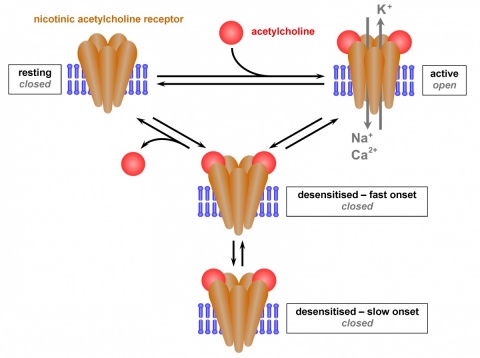
Im desensitisierten Zustand befindet sich der Agonist in der orthosterischen Bindungsstelle, aber es kommt zu keiner Aktivierung. Wie schon erwähnt, ist dies bei einem Agonist-Überschuss der Fall. Offensichtlich werden aber noch weitere (alloste-rische) Bindungsstellen belegt, die dann den desensitisierten Zustand stabilisieren. Diese „Desensitiser“ oder stille allosterische Modulatoren (SAM) genannten Substanzen können nicht nur Agonisten im Überschuss, sondern noch verschiedene andere Liganden sein. Welcher Mechanismus zugrunde liegt, ist noch unklar. Eine Hypothese ist, dass die Passage der Kationen an zwei Stellen geblockt werden kann, einerseits am „resting“ Tor und andererseits am „desensitisation“ Tor. Wird der nAChR aktiviert, müssen sich diese beiden Tore konzertiert öffnen. Bei einer Desensitisierung bleibt offensichtlich das „desensitisa-tion“ Tor geschlossen, so dass der Ionenkanal nicht mehr -ko-mplett offen ist, und demzufolge der intrinsische Effekt ausbleibt [38]. Auch die umgebende Membran scheint dabei eine Rolle zu spielen, welcher Funktionalitätszustand stabilisiert wird: Phosphatidylcholin-reiche Membranen stabilisieren den desensitisierten (geschlossenen, nicht mehr aktivierbaren) Zustand, während cholesterolreiche Membranen den „resting“ (geschlossenen, aktivierbaren) Zustand stabilisieren [39].
Allosterische Modulation des nAChR
Wie schon oben erwähnt, besitzt der nAChR nicht nur die orthosterischen Bindungsstellen für Agonisten bzw. Antagonisten, sondern auch allosterische Bindungsstellen. Definitionsgemäß ist die Wirkung allosterischer Modulatoren an den Effekt eines orthosterischen Agonisten gekoppelt: Entweder wird dessen Effekt verstärkt oder abgeschwächt. Positive allosterische Modulatoren (PAM) verstärken den Effekt (Affinität und/oder Aktivität) eines Agonisten, während negativ allosterische Modulatoren (NAM) den Effekt eines Agonisten abschwächen [40].
PAM werden in zwei Kategorien eingeteilt: Typ I PAM erhöhen den Effekt eines orthosterischen Agonisten, haben aber keinen Effekt auf die Desensitisierung des Rezeptors. Typ II PAM verringern die Eintrittswahrscheinlichkeit einer Desensitisierung und sind in der Lage, die intrinsische Aktivität vormals desensitisierter Rezeptoren wieder herzustellen [41, 42].
Aufgrund ihrer vielfältigen pharmakologischen Diversität bieten allosterische Modulatoren viele Möglichkeiten für therapeutische Ansätze [43]. Ein entscheidender Vorteil ist, dass ausschließlich der Effekt der orthosterischen Liganden moduliert wird; folglich sind dieses Zusammenspiel und seine Wirkung auf den Organismus nicht mehr gegeben, wenn einer der Partner ausfällt. Dieser „use-dependent“ Effekt ist gerade dann interessant, wenn die Gefahr besteht, dass bei zunehmender Dosierung der positive Effekt ins Negative umschlägt. Beim nAChR wäre dies beispielsweise bei Antagonisten der Fall. Die Population desensitisierter nAChR würde erniedrigt und die der aktivierbaren, aber geschlossenen („resting“) Zustände erhöht werden. Bei zu hohen Konzentrationen an Antagonisten würde die Population aber dermaßen in Richtung der „resting“ Zustände verschoben werden, dass kaum mehr offene Zustände vorhanden wären. Hinsichtlich des intrinsischen Effekts unterscheiden sich Antagonisierung und Desensitisierung nicht: In beiden Fällen kommt es zu keiner Durchleitung von Kationen. Bei einer durch Nervenkampfstoffe induzierten Desensitisierung der nAChR wäre die Gabe von Antagonisten allenfalls nur „das kleinere Übel“, und es wäre eine äußerst geringe therapeutische Breite zu erwarten. PAM wären daher die weitaus bessere Alternative [36].
nAChR-Modulatoren als neuer therapeutischer Ansatz
Um die therapeutische Lücke für die Behandlung von Vergiftungen mit Soman oder Tabun zu schließen, werden Wirkstoffe mit modulierendem Effekt auf die nAChR-Funktion benötigt. Insbesondere das „Einfrieren“ (auch als „open channel block“ beschrieben) des an sich metastabilen offenen Rezeptorzustands gewinnt an Bedeutung [44, 45]. Im Grunde genommen handelt es sich dabei um nichts anderes als die Wirkung von PAM am nAChR.
Frühere Versuche mit der Bispyridiniumverbindung SAD-128 (1,1’-Oxydimethylenbis(4-tert-butylpyridinium)dichlorid) zeigten einen therapeutischen Effekt gegen Soman in vitro und in vivo [46 - 49]. Da diese Verbindung keine Oxim-Gruppe besitzt und auch experimentell nachgewiesen wurde, dass der -Soman-AChE-Komplex nicht durch SAD-128 reaktiviert wurde, lag die Vermutung nahe, dass der positive therapeutische Effekt wohl einer direkten Interaktion mit nAChR zuzuschreiben war [50].
Bei MB327, einer dem SAD-128 strukturähnlichen Verbindung (1,1‘-(Propan-1,3-diyl)bis(4-tert-butylpyridinium)diiodid), wurde bewiesen, dass die Wiederherstellung der neuromuskulären Transmission nicht über die Reaktivierung des gehemmten Enzyms AChE, sondern rezeptorvermittelt abläuft [51 - 53]. An Muskeltyp-(2αβγδ)-nAChR, die aus dem elektrischen Organ des kalifornischen Zitterrochens (Torpedo californica) gewonnen wurden und dem humanen Subtyp sehr ähnlich sind, konnte gezeigt werden, dass MB327 nicht direkt mit der orthosterischen Bindungsstelle interagiert, wohl aber die Bindung ortho-sterischer Liganden wie beispielsweise des Agonisten Epibatidin beeinflusst [54, 55]. Die hierbei beobachtete Affinitätszunahme des Agonisten deutete auf einen allosterischen Effekt des MB327 hin. Unter Einsatz des gleichen nAChR-haltigen Materials wurden Funktionalitätsmessungen mit einer speziellen, am InstPharmToxBw entwickelten Bilayer-basierten Elektrophysiologie (sogenannte „zellfreie Elektrophysiologie“) durchgeführt. MB327 war in der Lage, nicht nur die Signale des Agonisten Carbamoylcholin zu verstärken, sondern auch eine Carbamoylcholin induzierte Desensitisierung wieder aufzuheben [56]. Damit war bewiesen, dass MB327 direkt mit dem nAChR interagiert und seine pharmakologische Wirkung als PAM entfaltet.
Allerdings hat MB327 zahlreiche Nachteile: Erstens liegt die Wirksamkeit nur im mikromolaren Bereich, so dass hohe Dosen appliziert werden müssen, was die praktische Anwendung erschwert. Zweitens ist die Wirkung von MB327 relativ unspezifisch [57]. Beispielsweise interagiert MB327 mit dem α7-nAChR in der gleichen Weise wie mit dem Muskeltyp-nAChR. Dies kann unter Umständen auch nachteilig sein, vor allem dann, wenn die nAChR der Atemmuskeln separat adressiert werden sollen. Beachtlich ist auch der antagonistische Effekt in muskarinischen Systemen. MB327 zeigte hierbei am glatten Muskel eine relaxierende Wirkung, wobei es sich um Konzentrationen handelte, welche bei mit Nervenkampfstoff vergifteten Atemmuskelpräparaten die Muskelfunktion wiederherstellten [58].
Rationales Wirkstoff-Design
Wenngleich MB327 eine zu geringe Wirkstärke aufweist, wurde dennoch eine Substanz identifiziert, die als Typ II PAM wirkt, d.h. vormals desensitisierte nAChR wieder in einen funktionalen Zustand überführt („resensitisiert“). Könnten auf dieser Basis Wirkstoffe entwickelt werden, die über eine für einen Arzneistoff ausreichend hohe Potenz verfügen und zudem auch die für einen therapeutischen Einsatz erforderlichen Kriterien erfüllen („drug-like properties“), so könnte damit ein Weg gefunden werden, die therapeutische Lücke bei der Behandlung von Vergiftungen – insbesondere mit Soman oder Tabun – zu schließen.
Dies war Ausgangspunkt für ein medizinalchemisches Projekt (E/U2AD/CF514/DF561) in Zusammenarbeit mit dem Arbeitskreis Prof. Dr. Wanner, Lehrstuhl für pharmazeutische / medizinische Chemie im Department Pharmazie – Zentrum für Pharmaforschung der LMU München, in dem unter Anwendung des rationalen Wirkstoffdesigns potente und selektive Modulatoren des nAChR entwickelt werden sollen.
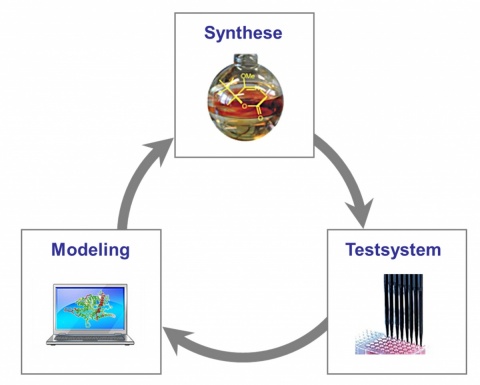
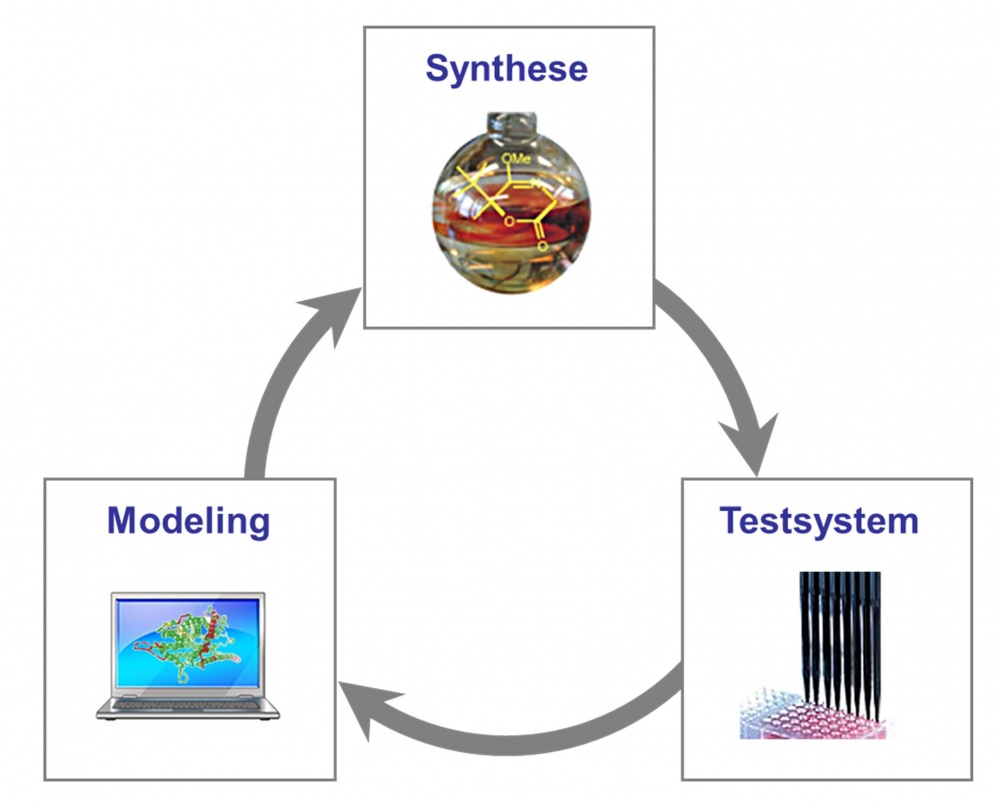
Dabei handelt es sich um einen iterativen Prozess aus Synthese, Affinitätsstudien mit massenspektrometrischer Detektion (MS--Bindungsstudien), Molecular Modeling, und elektrophysiologischen und physiologischen Untersuchungen (Abbildung 3).
Zu diesem Zweck wurden zahlreiche neue Bispyridiniumverbindungen mit unterschiedlichem Substitutionsmuster synthetisiert und anschließend pharmakologisch charakterisiert. Ausgewählte Verbindungen besitzen in Analogie zu MB327 eine 4-tert-Butylpyridinium-Grundstruktur und verfügen zusätzlich über eine weitere funktionelle Gruppe. Dadurch lässt sich der Einfluss des weiteren Substituenten im Sinne der Entwicklung von Struktur-Aktivitätsbeziehungen analysieren. Zusätzlich wurde eine neue, bis dato noch nicht beschriebene, effiziente und regioselektive Methode zur Synthese von 4-tert-Butylpyridinderivaten etabliert, welche als Basis für die Darstellung neuer Bispyridiniumsalze dient [59].
Um eine ausreichend empfindliche und zuverlässige Quantifizierung von MB327 in MS-Bindungsassays zu ermöglichen, wurde zunächst eine leistungsfähige LC-ESI[2])-MS/MS-Methode etabliert, die als internen Standard ein deuteriertes Analogon von MB327 verwendet. Anschließend wurde die Bindung von MB327 gegenüber dem Muskeltyp-nAChR in Sättigungsexperimenten, die zur Trennung von gebundenem und ungebundenem Marker einen Zentrifugationsschritt nutzen, charakterisiert. Hierfür wurden Membranpräparationen aus dem elektrischen Organ des kalifornischen Zitterrochens (Torpedo californica) eingesetzt, da ein adäquates Expressionssystem für den humanen Muskeltyp-nAChR bislang noch nicht verfügbar ist. Die Ergebnisse deuten auf eine sättigbare spezifische Bindung von MB327 hin und zeigen damit erstmals die Existenz von Bindungsstellen für dieses Bispyridiniumsalz. Mit dieser Methode konnte auch bestätigt werden, dass die Affinität von MB327 nur im mikromolaren Bereich liegt. Auf Basis der Sättigungsassays wurden danach Kompetitionsassays entwickelt, die das Screening der neu synthetisierten Substanzen auf ihre Affinität erlauben [61].
Für die computergestützte Suche nach möglichen Bindungsstellen von MB327 am nAChR wurde die Struktur des Torpedo--Muskeltyp-nAChR als Grundlage genommen, da dieser Rezeptorsubtyp (2α1ß1γδ) auch in den pharmakologischen Tests verwendet wird. Im Zuge des derzeit laufenden Projektes ist es gelungen, zwei mögliche, bis dato unbekannte Bindungsstellen erstmalig zu identifizieren. Diese Ergebnisse konnten auch in Homologiemodellen des humanen adulten Muskeltyp-nAChR erzielt werden. Darüber hinaus zeigte es sich, dass die beiden möglichen Bindungsstellen auch sehr gut von unsymmetrisch substituierten Bispyridiniumverbindungen mit polaren Substituenten adressiert werden könnten.
Auch wenn die bisher entwickelten in silico Methoden bereits sehr leistungsfähig sind, so ist es erforderlich, sie regelmäßig mit neuen Ergebnissen aus der Affinitätsbestimmung (MS-Bindungsstudien) und pharmakologischen Untersuchungen zu aktualisieren und sie so kontinuierlich weiter zu verbessern. Die auf diese Weise zugänglichen, besonders aussagekräftigen Modelle leisten wertvolle Dienste bei der weiteren Optimierung der Modulatoren, wie auch bei der Identifizierung ihrer Target-Bindungsstellen.
Pharmakologische Untersuchungen
Die pharmakologischen Untersuchungen wurden am InstPharmToxBw durchgeführt. Dabei handelte es sich um elektrophysiologische (automatisierte Patch-Clamp-Technik an ganzen Zellen, Bilayer-basierte Elektrophysiologie unter Verwendung von Plasmamembranpräparationen mit Muskeltyp--nAChR) und physiologische Methoden (Muskelkraftmessungen an Zwerchfellpräparationen der Ratte).
Die Patch-Clamp-Experimente wurden mit CHO[3]) Zellen, die den humanen nAChR des Subtyps α7 stabil exprimieren, durchgeführt. Auch wenn es sich um einen anderen nAChR-Subtyp handelt als den Muskeltyp-nAChR, geben diese Experimente äußerst wertvolle Hinweise auf den intrinsischen Effekt neuer Substanzen. Hierbei konnten weitere Verbindungen identifiziert werden, die eine Aktivierung der nAChR durch die Stabilisierung des aktiven Zustands aufrecht erhielten und den Eintritt der durch Nikotin-Überschuss verursachten Desensitisierung durch Verlängerung der Öffnungsdauer verhinderten. Diese Effekte wurden nur in Anwesenheit des Agonisten Nikotin induziert, was auf eine positive allosterische Modulation hindeutet. Auch in diesem System lagen die Aktivitäten der neuen Substanzen nur im mikromolaren Bereich. Nichtsdestotrotz konnten erste Struktur-Wirkungsbeziehungen erkannt werden, die im Kontext mit den weiteren molekularen und physiologischen Methoden (Bilayer-basierte Elektrophysiologie, Kraftmessungen an Muskelbiopsaten) essentiell sind [62].
Für die physiologischen Untersuchungen der neuen Substanzen wurde als Gewebemodell die Zwerchfellpräparation der Ratte eingesetzt.
Hierbei wurden Zwerchfellhemisphären in einem Multiorganbadsystem an Myographen fixiert und mittels indirekter elektrischer Feldstimulation (20, 50, und 100 Hz) zur Kontraktion gebracht. Die Muskelkraft wurde vor und nach Soman-Gabe sowie nach Applikation verschiedener Bispyridiniumverbindungen (1 - 300 µM[4])) gemessen und als Zeit-Kraft-Integral ausgewertet.
Soman, in der Konzentration 3 µM, führte zur vollständigen Hemmung der Muskulatur. Auch nach Auswaschen des Organophosphats konnte die Muskelkraftentwicklung nicht wieder hergestellt werden. Mit MB327 konnte bei der niedrigen Stimulationsfrequenz von 20 Hz bei einer Konzentration von 300 µM eine Wiederherstellung der Muskelkraft um etwa 30 % beobachtet werden. Auch weitere Bispyridiniumverbindungen waren in der Lage, wieder Muskelkontraktionen nach einer -Soman-Vergiftung hervorzurufen. Wie bei den Patch-Clamp--Versuchen hing dies stark vom Substitutionsmuster ab. Allerdings war auch in diesem Modell die Wirkung der Bispyridi-niumverbindungen erst bei relativ hohen Konzentrationen (100 - 300 µM) zu beobachten. Bei höheren Stimulationsfrequenzen und damit einer vermehrten Ausschüttung des Neurotransmitters Acetylcholin nahm ihre Wirksamkeit wieder sehr stark ab.
In Summe konnten relativ deutliche Korrelationen zu den elektrophysiologischen Methoden (Patch Clamp, Bilayer-basierte Elektrophysiologie) beobachtet werden [63]. Alle hier gewonnenen Kenntnisse über Struktur-Wirkungsbeziehungen vervollständigten zudem die Datenlage für das in silico Screening.
Ausblick
Die Applikation von Wirkstoffen, welche die Aktivität desensitisierter nAChR trotz akkumuliertem Acetylcholin im synaptischen Spalt wiederherstellen, könnte ein möglicher therapeutischer Ansatz bei konventionell mit Atropin und Oximen nicht therapierbaren Nervenkampfstoffvergiftungen sein. Hierzu sind Substanzen mit Typ II PAM-Eigenschaften, die erstmalig für MB327 in verschiedenen pharmakologischen Untersuchungen nachgewiesen wurden, in der Lage. Kürzlich konnte in MS-Bindungsstudien gezeigt werden, dass sich die Bindung von MB327 am nAChR absättigen lässt, d.h. für MB327 explizite Bindungsstellen existieren. Mit Hilfe von in silico Computermodellen konnten zwei mögliche Bindungsstellen identifiziert und optimale Pharmakophore für die Adressierung dieser Bindungsstellen berechnet werden.
Als nächster Schritt sind Substanzen zu synthetisieren, die auf Grundlage der theoretisch bestimmten Pharmakophore entworfen und dann in die verschiedenen pharmakologischen Testungen eingeschleust werden. Die daraus gewonnenen Ergebnisse komplettieren immer weiter die Datenlage für umfassende Struktur-Wirkungsbeziehungen.
Die stetige Verbreiterung der Datenbasis hinsichtlich der Beziehungen zwischen chemischer Struktur und biologischer Aktivität wird es erlauben, die erzeugten Computer-Modelle zum -Ligand- und Struktur-basierten Design, zum Nachweis der -Target-Bindungsstellen und zum in silico Screening deutlich zu verbessern. Dies wird entscheidend zur Identifizierung und zum Design neuer Wirkstoff-Derivate und Wirkstoff-Klassen beitragen.
Da hierfür bereits wegweisende Ergebnisse erzielt worden sind, sollte diese Entwicklung mit dem Ziel, potentere und selektivere Wirkstoff zu finden, unbedingt weiter verfolgt werden.
Schlussfolgerung
Obwohl nAChR eine wichtige Rolle bei Nervenkampfstoffvergiftungen spielen, sind sie bislang als mögliche Zielstrukturen für eine therapeutische Intervention vernachlässigt worden. Dies lag wohl auch darin begründet, dass nur konventionelle nAChR Antagonisten betrachtet wurden, bei denen aber die Gefahr einer geringen therapeutischen Breite besteht.
Ein vielversprechender Ansatz könnten Typ II PAM sein, die desensitisierte nAChR wieder in einen funktionalen Zustand versetzen – d.h. als „Resensitiser“ wirken – wobei die Wirkung nicht autark, sondern in Abhängigkeit mit dem Agonisten erfolgt.
Die Bispyridiniumverbindung MB327 wurde als ein solcher Typ II PAM identifiziert, weist aber noch eine zu geringe -Wirkstärke und Selektivität auf. Nichtsdestotrotz stellt diese Substanz eine wertvolle Grundlage für die Entwicklung potenterer Wirkstoffe dar. Denn, basierend auf MB327 als Leitstruktur, können neue Wirkstoffkandidaten synthetisiert und pharmakologisch charakterisiert werden. Die kürzlich erfolgte Identifizierung möglicher Bindungsstellen des Bispyridiniumsalzes MB327 am nAChR kann nun für die Verfeinerung des Pharmakophors genutzt werden und bietet die Möglichkeit, gänzlich neue Perspektiven in der Antidotforschung zu eröffnen. Gerade im Hinblick auf die jüngsten Ereignisse in Syrien wiegt die therapeutische Lücke bei der Behandlung von Soman- oder -Tabun-Vergiftungen schwer. Umso wichtiger ist, dass der mit diesem Forschungsprojekt beschrittene Weg weiter mit Hochdruck verfolgt wird.
Literatur
- MacPhee-Quigley K, Taylor P, Taylor S: Primary structures of the catalytic subunits from two molecular forms of acetylcholinesterase. A comparison of NH2-terminal and active center sequences. J. Biol. Chem. 1985; 260(22): 12185 - 12189.
- Taylor P, Radic Z, Hosea NA, Camp S, Marchot P, Berman HA: Structural bases for the specificity of cholinesterase catalysis and inhibition. Toxicol. Lett. 1995; 82 - 83(0): 453 - 458.
- Molenaar PC, Oen BS, Polak RL, van der Laaken AL: Surplus acetylcholine and acetylcholine release in the rat diaphragm. J Physiol. 1987; 385: 147 - 367.
- Gunderson CH, Lehmann CR, Sidell FR, Jabbari B: Nerve agents: A review. Neurology. 1992; 42(5): 946 - 50.
- Marrs TC: Organophosphate poisoning. Pharmacol. Ther. 1993; 58(1): 51 - 66.
- Grob D: Manifestations and treatment of nerve gas poisoning in man. U. S. Armed Forces Med J. 1956; 7(6): 781 - 789.
- Rickett DL, Glenn JF, Beers ET: Central respiratory effects versus neuromuscular actions of nerve agents. Neurotoxicology. 1986; 7(1): 225 - 236.
- Newmark J: Therapy for nerve agent poisoning. Arch. Neurol. 2004; 61(5): 649 - 652.
- Thiermann H, Szinicz L, Eyer F, Worek F, Eyer P, Felgenhauer N, Zilker T: Modern strategies in therapy of organophosphate poisoning. Toxicol. Lett. 1999; 107(1 - 3): 233 - 239.
- Thiermann H, Szinicz L, Eyer P, Felgenhauer N, Zilker T, Worek F: Lessons to be learnt from organophosphorus pesticide poisoning for the treatment of nerve agent poisoning. Toxicology. 2007; 233(1 - 3): 145 - 154.
Für die Verfasser:
Oberfeldapotheker Karin V. Niessen
Institut für Pharmakologie und Toxikologie der Bundeswehr
Neuherbergstr. 11, 80937 München
E-Mail: karinniessen@bundeswehr.org
[1])
LC-MS/MS = Flüssigchromatographie gekoppelt mit Tandem-Massenspektrometrie
[2]) ESI = Elektrospray-Ionisation
[3])
CHO = Chinese Hamster Ovary; vielseitig verwendete Zellkultur-linie, die aus den Ovarien der chinesischen Hamsterart Cricetulus griseus gewonnen wurde
[4]) µM = µmol/l
Datum: 01.06.2017
Quelle: Wehrmedizinische Monatsschrift 2017/5









{kind=link}
{kind=link}
{kind=link}